
Introduction
Atrial fibrillation (AF) remains the most common sustained cardiac arrhythmia encountered in clinical practice, characterized by disorganized atrial electrical activity, loss of coordinated contraction, and an irregularly irregular ventricular response. Its global burden is vast — affecting more than 37 million individuals worldwide — and its complications, including stroke and heart failure, render it a significant public health concern. Traditionally, atrial fibrillation has been attributed to a triad of structural heart disease, systemic comorbidities, and advancing age. However, emerging evidence has brought renewed attention to a lesser-known yet clinically relevant phenomenon: drug-induced atrial fibrillation (DIAF).
While clinicians remain vigilant about proarrhythmic risks such as torsades de pointes, the potential for pharmacological agents to provoke atrial fibrillation is often overlooked. This omission is not merely academic — it has direct consequences in clinical safety, therapeutic decision-making, and pharmacovigilance. The recent narrative review on this topic highlights how a wide array of medications — spanning from cardiovascular agents and bronchodilators to chemotherapeutic and psychotropic drugs — may precipitate AF through diverse electrophysiological and systemic pathways.
This article revisits the forgotten concept of drug-induced atrial fibrillation, examines its mechanistic underpinnings, outlines the culpable drug classes, and proposes strategies for recognition, prevention, and clinical management. The intention is not to vilify essential pharmacotherapies, but to cultivate the nuanced awareness required for their safe use in vulnerable cardiac terrain.
The Forgotten Adverse Effect: Why DIAF Slipped Through the Cracks
In contrast to well-characterized ventricular arrhythmias, atrial proarrhythmia has historically received little formal recognition in pharmacology. Part of this neglect stems from diagnostic ambiguity: unlike QT prolongation or ventricular tachyarrhythmia, which are quantifiable and visually striking on electrocardiograms, atrial fibrillation represents a multifactorial, less predictable event. It may emerge transiently, resolve spontaneously, and therefore evade causative attribution to drugs.
Additionally, the latency period between drug exposure and arrhythmia onset varies widely — from hours to weeks — further obscuring temporal associations. The polypharmacy common in modern medicine compounds the issue, as multiple agents may contribute to arrhythmic risk synergistically through overlapping mechanisms, such as sympathetic activation or electrolyte imbalance.
Regulatory frameworks have likewise lagged in mandating systematic assessment of atrial arrhythmogenic potential during drug development. The focus of early safety pharmacology studies, guided by ICH E14 and S7B guidelines, has been ventricular repolarization (QT interval prolongation) rather than atrial electrophysiology. Consequently, many agents entered the market without rigorous screening for atrial arrhythmogenic effects.
In short, drug-induced AF became the forgotten arrhythmia—hidden in plain sight, clinically significant yet insufficiently studied, its recognition largely dependent on astute clinicians and case report accumulation rather than structured trials.
Mechanisms of Drug-Induced Atrial Fibrillation: The Multilevel Pathophysiology
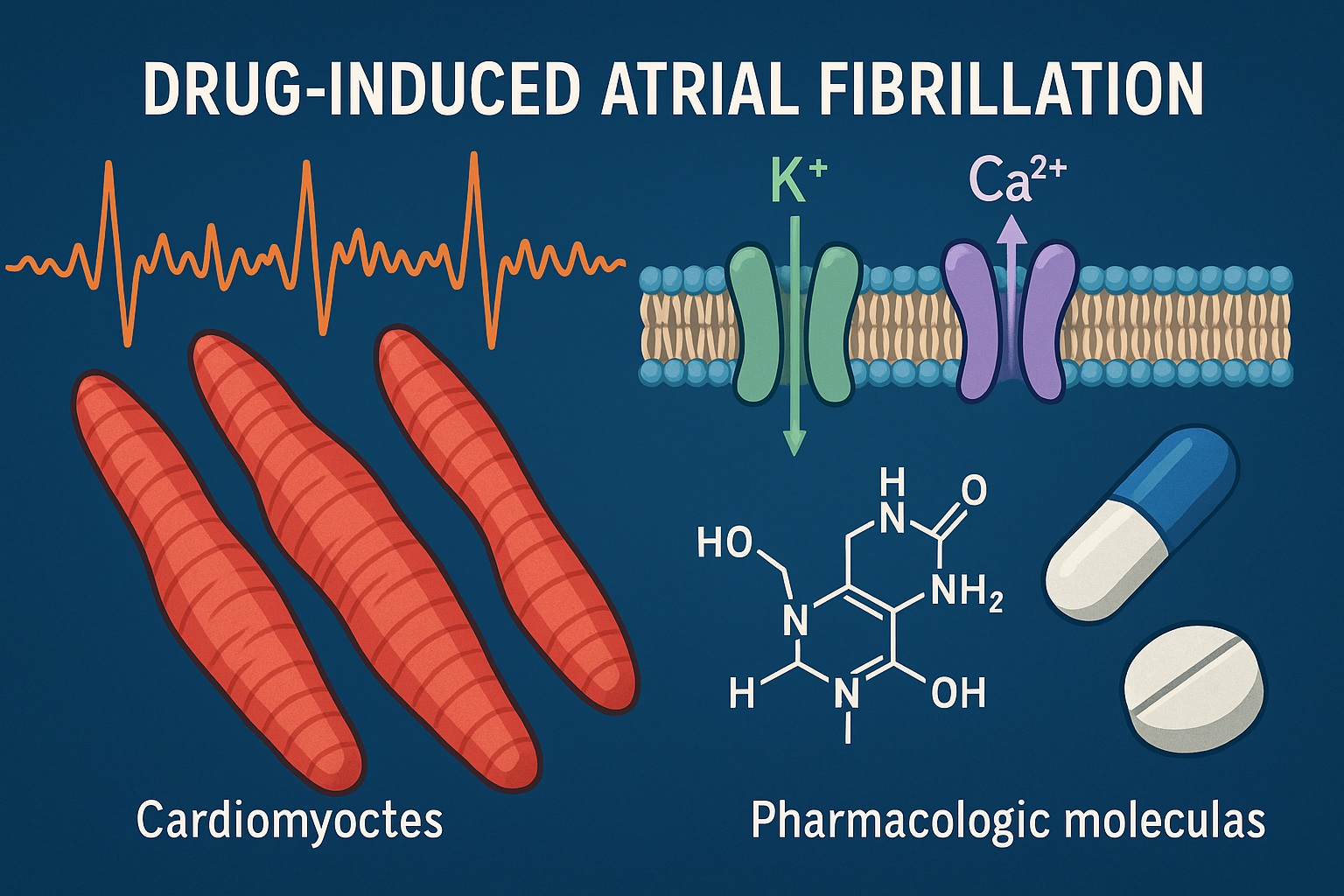
The pathogenesis of DIAF is complex, involving electrophysiological, neurohumoral, and structural components. No single unifying mechanism explains all cases; rather, each drug may trigger AF through a distinct constellation of effects.
1. Electrophysiological Disturbances
Certain drugs alter atrial refractory periods, conduction velocity, or automaticity. Sympathomimetic agents such as β₂-adrenergic agonists increase intracellular cyclic AMP, enhancing calcium influx and predisposing to triggered activity. Similarly, drugs that inhibit potassium channels can shorten atrial action potentials and promote re-entry phenomena, particularly in atria already susceptible to conduction heterogeneity.
Paradoxically, some antiarrhythmic drugs themselves—especially class I agents like flecainide or propafenone—can provoke AF by uneven conduction slowing across atrial tissue, facilitating re-entrant circuits. The electrophysiologic substrate thus becomes proarrhythmic, not protective, when applied inappropriately or in structurally abnormal atria.
2. Neurohumoral Activation
Drugs that stimulate sympathetic tone or suppress vagal activity destabilize the autonomic balance controlling atrial excitability. For instance, β₂-agonists, thyroid hormone analogues, and corticosteroids increase sympathetic drive, while agents like atropine remove vagal restraint. The resulting adrenergic dominance shortens atrial refractory periods and enhances automaticity.
Conversely, abrupt withdrawal of β-blockers or clonidine can cause rebound sympathetic surges, precipitating AF in predisposed individuals. The dynamic interplay between adrenergic and parasympathetic systems explains why both stimulation and withdrawal phenomena can yield similar arrhythmic outcomes.
3. Structural and Metabolic Effects
Atrial remodeling, ischemia, or inflammation induced by drugs create the substrate upon which electrical triggers act. Chemotherapeutic agents such as anthracyclines, cyclophosphamide, and cisplatin may cause direct cardiomyocyte injury, oxidative stress, and interstitial fibrosis, all of which increase atrial vulnerability. Similarly, electrolyte disturbances secondary to diuretics or laxatives — hypokalemia, hypomagnesemia — facilitate ectopic atrial firing.
Finally, drugs causing thyrotoxicosis (e.g., amiodarone) or systemic inflammation (e.g., interferons) alter myocardial metabolism and ion channel expression, completing the pathological triad: trigger, substrate, and modulator.
The Usual Suspects: Drug Classes Implicated in Atrial Fibrillation
A striking aspect of DIAF is its pharmacological diversity. From respiratory drugs to psychotropics, the list of offenders transcends specialty boundaries. Understanding these categories allows clinicians to contextualize AF within pharmacotherapy rather than serendipity.
Cardiovascular Drugs
Ironically, medications intended to protect the heart can, under specific conditions, destabilize it. Antiarrhythmic drugs—particularly class IC agents—are known culprits, especially in patients with structural heart disease. Excessive sodium channel blockade slows intra-atrial conduction, creating conditions conducive to re-entry.
Diuretics and vasodilators can precipitate AF indirectly through electrolyte depletion or reflex sympathetic activation. Even digoxin, though once a cornerstone of rate control, may induce atrial ectopy at toxic concentrations by enhancing intracellular calcium.
Sympathomimetics and Bronchodilators
β₂-agonists (e.g., salbutamol, terbutaline) are frequent offenders, especially when administered at high doses in acute exacerbations of asthma or COPD. Their action increases intracellular cAMP and calcium, generating afterdepolarizations that may trigger AF. The risk is amplified when combined with theophylline, another agent notorious for lowering the atrial fibrillation threshold via adenosine receptor antagonism.
Thyroid Hormones and Corticosteroids
Excess thyroid hormone, whether endogenous or iatrogenic, accelerates cardiac conduction and enhances adrenergic receptor sensitivity. The resulting hypermetabolic state predisposes to AF in up to 15% of hyperthyroid patients. Similarly, high-dose corticosteroids promote AF by causing hypokalemia, fluid retention, and sympathetic overactivity — the classic “steroid arrhythmia.”
Chemotherapeutic and Targeted Agents
Cancer therapeutics represent a growing frontier in cardiotoxicity. Anthracyclines, cyclophosphamide, 5-fluorouracil, and cisplatin have long been linked to arrhythmias via oxidative myocardial injury. More recently, tyrosine kinase inhibitors (TKIs) and immune checkpoint inhibitors have emerged as sources of inflammation-mediated atrial arrhythmogenesis. The incidence of AF with ibrutinib, a Bruton’s tyrosine kinase inhibitor, reaches 10–15%, necessitating vigilant cardiac monitoring during therapy.
Psychotropic and Central Nervous System Agents
Certain tricyclic antidepressants and antipsychotics (notably clozapine and ziprasidone) alter cardiac conduction through sodium and potassium channel blockade. Additionally, stimulants such as amphetamines or methylphenidate elevate sympathetic tone, while selective serotonin reuptake inhibitors may influence platelet aggregation and endothelial function—factors that indirectly modulate atrial electrophysiology.
Others: Hidden Contributors
Agents as disparate as interferon-α, erythropoietin, and phosphodiesterase inhibitors have been associated with AF episodes. Even over-the-counter sympathomimetic decongestants (pseudoephedrine) can trigger transient AF in sensitive individuals, particularly when coupled with caffeine or stress.
Risk Factors and Patient Susceptibility
Drug exposure alone rarely explains atrial fibrillation onset; host susceptibility defines the boundary between tolerance and toxicity. The review identifies several predisposing factors:
- Advanced age, with age-related atrial fibrosis and conduction heterogeneity.
- Pre-existing cardiac disease, such as hypertension, ischemia, or valvular abnormalities.
- Electrolyte disturbances or renal dysfunction affecting drug clearance.
- Polypharmacy, increasing the likelihood of pharmacodynamic and pharmacokinetic interactions.
- Inflammatory or hyperadrenergic states, including infections, trauma, or thyroid disorders.
Elderly polymedicated patients—precisely those most exposed to chronic pharmacotherapy—thus form the epidemiological epicenter of DIAF. Awareness in this population is paramount, as symptoms may be attributed to “aging” rather than drug effect, delaying recognition and intervention.
The Clinical Presentation: Recognizing the Unrecognized
Drug-induced AF is clinically indistinguishable from other forms of atrial fibrillation, yet subtle temporal cues may hint at pharmacological causation. Onset soon after drug initiation, dose escalation, or interaction with a new agent should raise suspicion. Transient, self-limiting AF following intravenous or high-dose therapy—especially in patients without prior arrhythmic history—further strengthens causality.
The Naranjo Adverse Drug Reaction Probability Scale and similar causality assessment tools can help formalize clinical reasoning, though they remain imperfect in complex scenarios. Importantly, withdrawal or dose reduction of the offending drug often leads to spontaneous rhythm normalization—a diagnostic clue in itself.
Laboratory evaluation should include electrolyte panels, thyroid function tests, and drug level measurements where applicable. Continuous ECG monitoring, particularly in hospitalized or oncology patients receiving high-risk medications, enhances detection of asymptomatic or paroxysmal episodes.
Diagnostic Challenges: Attribution in a Polypharmacy Era
Assigning causality in DIAF is inherently difficult. In modern clinical environments, patients often take five to ten concurrent medications, many with overlapping cardiovascular effects. Moreover, atrial fibrillation may manifest weeks or even months into therapy, obscuring temporal associations. Drug–disease interactions further complicate interpretation—for example, β₂-agonists in a COPD patient whose hypoxia alone predisposes to AF.
Causality grading therefore requires a multidimensional assessment, integrating temporal relationships, biological plausibility, exclusion of alternative causes, and response to withdrawal. In research, controlled rechallenge may confirm diagnosis, but ethically, such strategies are seldom justified in clinical practice. Hence, DIAF remains largely a diagnosis of probability, not certainty, underscoring the need for clinician vigilance and robust pharmacovigilance reporting systems.
Prevention and Clinical Management
The cornerstone of DIAF management is prevention through awareness. Recognizing drugs with arrhythmogenic potential allows clinicians to anticipate risk and implement proactive monitoring strategies.
Key preventive principles include:
- Baseline cardiac assessment before initiating high-risk drugs, including ECG and electrolyte evaluation.
- Dose optimization — using the lowest effective dose to minimize systemic stress.
- Monitoring during titration, especially in elderly or comorbid patients.
- Avoiding pharmacodynamic interactions, such as combining sympathomimetics with theophylline or corticosteroids.
Once AF develops, the standard management principles apply: rate or rhythm control, anticoagulation based on CHA₂DS₂-VASc scoring, and elimination of the precipitating drug. In many cases, sinus rhythm restores spontaneously following drug withdrawal, obviating the need for aggressive intervention.
Pharmacological reversal is preferred over invasive measures unless hemodynamic compromise ensues. Importantly, clinicians should resist the reflex of adding another antiarrhythmic agent to treat DIAF without first discontinuing the offender—a classic pitfall of “prescribing cascades.”
Pharmacovigilance and Regulatory Implications
Despite growing awareness, reporting of DIAF remains sporadic. Few randomized controlled trials have systematically captured atrial arrhythmias as predefined endpoints. This underreporting perpetuates the illusion of rarity and hinders risk quantification.
Regulatory agencies are beginning to acknowledge the gap. Post-marketing surveillance databases, such as FAERS (FDA Adverse Event Reporting System) and EudraVigilance, now allow specific categorization of atrial fibrillation as an adverse event. However, spontaneous reporting suffers from under-detection, incomplete documentation, and lack of denominator data.
To bridge this gap, integration of electronic health records (EHRs) with pharmacovigilance analytics and machine learning algorithms could detect emerging signals earlier. Prospective registries focusing on cardio-oncology and geriatric pharmacology may also enrich real-world data. Ultimately, formal inclusion of atrial arrhythmogenic potential in drug development safety screening is overdue—a reform that could mirror the success of QT interval monitoring in preventing torsadogenic agents from reaching patients.
Lessons for Clinicians: Rational Prescribing in a Vulnerable Population
The clinical takeaway from the narrative review is clear: any drug can be arrhythmogenic under the wrong circumstances. Physicians must therefore adopt a contextual prescribing mindset, weighing not only therapeutic efficacy but also electrophysiological compatibility.
In practice, this entails:
- Taking thorough medication histories, including over-the-counter and herbal products.
- Recognizing that new-onset AF in a patient on multiple medications should trigger a drug review before assuming idiopathic etiology.
- Educating patients on recognizing palpitations, dizziness, or dyspnea early during therapy.
- Collaborating with pharmacists and cardiologists to evaluate complex regimens for arrhythmic risk.
In doing so, clinicians can transform what is currently a “forgotten adverse effect” into a preventable complication through vigilance, communication, and evidence-based pharmacovigilance.
Conclusion
Drug-induced atrial fibrillation is neither rare nor trivial — it is merely under-recognized. The phenomenon embodies the intricate intersection of pharmacology, electrophysiology, and patient vulnerability. The reviewed evidence underscores that AF may arise not only from disease but from the very drugs designed to heal, through mechanisms ranging from ion channel modulation and sympathetic activation to inflammation and oxidative stress.
As medicine evolves toward polytherapy and molecular targeting, the potential for unintended cardiac consequences inevitably expands. The path forward demands a dual commitment: scientific rigor in drug safety evaluation and clinical mindfulness in prescribing practices. Recognizing DIAF is not about limiting therapeutic innovation—it is about ensuring that pharmacological progress does not come at the expense of rhythm stability.
In the end, the forgotten arrhythmia deserves remembrance—not as a relic of pharmacovigilance negligence, but as a reminder that every therapeutic advance carries within it the duty of vigilance.
FAQ: Drug-Induced Atrial Fibrillation
1. Which drugs are most frequently implicated in atrial fibrillation?
The most consistently reported culprits include β₂-agonists (salbutamol, terbutaline), theophylline, corticosteroids, thyroid hormone, anthracyclines, cyclophosphamide, and tyrosine kinase inhibitors such as ibrutinib. Antiarrhythmic agents, particularly class I drugs, can also paradoxically trigger AF in susceptible patients.
2. How can clinicians differentiate drug-induced AF from other forms?
Temporal association with drug initiation, dose escalation, or combination therapy provides key clues. Resolution upon withdrawal strongly supports causality. Exclusion of alternative triggers—electrolyte imbalance, infection, or ischemia—is essential for accurate attribution.
3. Should all patients receiving high-risk drugs undergo cardiac monitoring?
Not universally, but targeted monitoring is recommended for high-risk groups: elderly individuals, patients with structural heart disease, electrolyte disturbances, or concurrent use of multiple proarrhythmic agents. Baseline ECG and follow-up during dose titration enhance safety and early detection.