

Introduction
In oncology, serendipity often guides discovery more effectively than design. Aspirin’s anticancer potential was once a curiosity; today, it’s a clue to something deeper—a pharmacological paradox hiding inside an old drug class. Among nonsteroidal anti-inflammatory drugs (NSAIDs), sulindac stands as a particularly intriguing case. It has shown the ability to suppress colorectal adenomas, yet its long-term clinical use is crippled by the very mechanism that made it famous: cyclooxygenase (COX) inhibition.
This conflict has motivated decades of research to untangle sulindac’s true antineoplastic mechanism. A mounting body of evidence now points toward an unexpected molecular pathway—the cyclic guanosine monophosphate (cGMP)/protein kinase G (PKG) axis. When properly activated, this signaling cascade appears capable of reversing oncogenic signaling downstream of APC mutations, the genetic hallmark of most colorectal cancers.
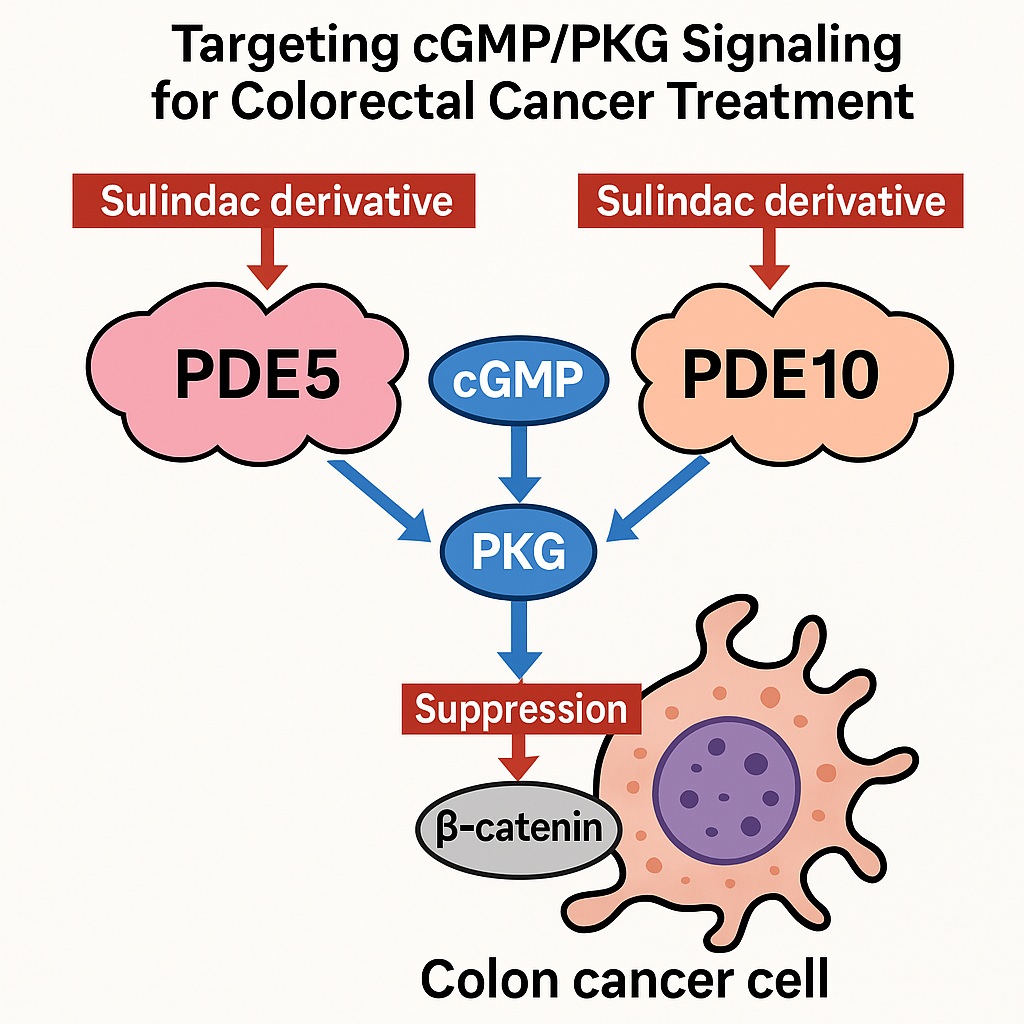
The latest frontier in this story involves novel sulindac derivatives that have been deliberately stripped of COX-inhibitory activity, yet retain—and even enhance—their ability to inhibit tumor cell proliferation. These compounds target phosphodiesterases (PDEs), particularly PDE5 and PDE10, thereby restoring cGMP homeostasis and reactivating PKG signaling to suppress the Wnt/β-catenin pathway.
The result is a reimagined approach to cancer prevention—one that reframes an old anti-inflammatory drug as a selective molecular weapon against the earliest events in colorectal tumorigenesis.
From Polyps to Pathways: The Burden of Colorectal Cancer and FAP
Colorectal cancer remains one of the most preventable malignancies in modern medicine. In the United States alone, roughly 28 million individuals harbor precancerous colonic adenomas. For many, these lesions never progress. But for others—particularly those carrying inherited mutations in the APC gene—the story ends differently.
Familial adenomatous polyposis (FAP), though rare, epitomizes this high-risk state. It’s an autosomal dominant syndrome that predisposes patients to develop hundreds or even thousands of adenomatous polyps by adolescence. Without surgical intervention, colorectal carcinoma is nearly inevitable by middle age.
Current management relies on repeated colonoscopic surveillance and, ultimately, colectomy—strategies that are effective but hardly elegant. They treat the symptom, not the molecular cause.
This is where pharmacologic intervention could, in principle, change everything. A drug capable of safely suppressing adenoma formation or delaying malignant transformation would spare countless patients from surgical mutilation. Unfortunately, existing NSAIDs—though effective in reducing adenoma burden—carry serious cardiovascular, renal, and gastrointestinal risks tied to COX inhibition. Hence, a new generation of NSAID-like molecules is being engineered to preserve the anticancer benefits while shedding the toxic baggage.
When COX Isn’t the Culprit: Decoding the NSAID Paradox
The idea that NSAIDs prevent cancer primarily through COX inhibition has long been comfortable but incomplete. Epidemiological data support NSAID use in lowering colorectal cancer incidence by nearly 50%, but pharmacological inconsistencies abound.
For one, the rank order of antitumor potency among NSAIDs doesn’t match their capacity to inhibit prostaglandin synthesis. Moreover, non-COX inhibitory metabolites like exisulind (sulindac sulfone) retain anticancer efficacy despite lacking any measurable impact on prostaglandins. Such findings demolish the simplistic COX-centric model and redirect attention to COX-independent mechanisms.
Research eventually converged on cyclic nucleotide signaling—specifically, the role of phosphodiesterases in modulating intracellular cGMP. Sulindac and its metabolites, it turns out, inhibit certain PDE isozymes, resulting in elevated cGMP levels and PKG activation, which in turn suppress oncogenic β-catenin signaling.
This realization marks a critical pivot. The true antineoplastic effect of sulindac may have nothing to do with inflammation at all—it may be about restoring lost cyclic nucleotide signaling balance in cells hijacked by cancer.
Engineering the Perfect Derivative: The Rise of COX-Free Sulindac Analogs
The next logical step was chemical redesign. Using molecular modeling, researchers sought to “design out” COX binding from sulindac’s structure while retaining, or enhancing, its antiproliferative potency.
The breakthrough came with sulindac sulfide amide (SSA)—a modified analog where the carboxylic acid group critical for COX interaction was replaced with a dimethyl ethyl amide moiety. The result was transformative: SSA lacked COX-1 and COX-2 inhibitory activity but retained potent anticancer effects in cell lines and animal models.
From there, medicinal chemists embarked on an exhaustive combinatorial synthesis campaign, generating over 1,500 sulindac derivatives with variations across the indene scaffold. Each compound was rigorously tested for:
- Growth inhibition in colon cancer cell lines (HT29, HCT116, SW480).
- Selectivity against normal colonocytes (NCM460).
- PDE isozyme inhibition profiles and cGMP modulation.
Through this systematic process, researchers identified compounds with enhanced potency and selectivity, pinpointing a shared mechanism: inhibition of PDE5 and PDE10, leading to cGMP elevation and β-catenin suppression.
PDE5: An Unexpected Cancer Target
Historically, PDE5 has been associated with smooth muscle relaxation and vascular tone regulation—famously targeted by drugs like sildenafil. But in colon cancer, its role appears far less benign.
Colon adenomas and adenocarcinomas overexpress PDE5, while normal colon tissue expresses little to none. This makes PDE5 not just a biomarker, but a therapeutic vulnerability.
Sulindac derivatives demonstrated strong PDE5 inhibition, leading to a cascade of molecular events:
- Accumulation of cGMP,
- Activation of PKG,
- Phosphorylation and degradation of β-catenin, and
- Suppression of Tcf/Lef transcriptional activity—the engine of tumor proliferation.
In contrast, conventional PDE5 inhibitors like sildenafil require impractically high concentrations to elicit similar effects in vitro, likely due to efflux mechanisms in tumor cells. Nevertheless, in animal models, PDE5 inhibition has shown the ability to suppress inflammation-driven colon tumorigenesis and possibly enhance antitumor immunity by mitigating myeloid-derived suppressor cell activity.
Thus, while PDE5 may not be the sole target, it’s an undeniably important one—a gatekeeper of cyclic signaling that cancer cells exploit to sustain growth.
PDE10: The Silent Partner in Tumor Proliferation
If PDE5 is the door, PDE10 may be the lock. Originally known for its role in dopaminergic signaling in the brain, PDE10’s expression in peripheral tissues is negligible—except, curiously, in colon tumors.
Studies have shown that PDE10 is strongly upregulated in adenomas and adenocarcinomas, but nearly absent in normal colonic mucosa. Genetic knockdown or pharmacologic inhibition of PDE10 selectively impairs cancer cell viability while sparing normal cells—a hallmark of an ideal therapeutic target.
Mechanistically, PDE10 also degrades cGMP, and its inhibition mirrors that of PDE5, leading to PKG activation and suppression of β-catenin-driven transcription. Importantly, this pathway directly counters the oncogenic consequences of APC mutations, which disrupt β-catenin regulation and promote uncontrolled proliferation.
The low physiological expression of PDE10 outside the central nervous system provides an additional advantage: targeting PDE10 could minimize systemic toxicity, offering a new degree of precision in chemoprevention.
Why Not Both? The Case for Dual PDE5/10 Inhibition
In reality, cancer signaling rarely hinges on a single molecule. PDE5 and PDE10 often coexist in colon cancer cells, working in concert to deplete cGMP and silence PKG. This functional redundancy inspired a new hypothesis: dual inhibition might yield synergistic anticancer effects.
Indeed, experimental evidence supports this view. Simultaneous blockade of PDE5 and PDE10 produces additive suppression of tumor cell growth, far exceeding the effect of either inhibitor alone. Dual inhibitors sustain cGMP elevation longer, ensuring persistent PKG activation and more thorough degradation of oncogenic β-catenin.
Moreover, combining targets could bypass resistance mechanisms, such as compensatory upregulation of alternative PDE isozymes. This elegant strategy promises broader efficacy without added toxicity, since neither enzyme is significantly expressed in vital normal tissues.
In essence, dual PDE5/10 inhibition represents a molecular pincer attack—disabling cancer’s redundant cyclic signaling systems with surgical precision.
The Molecular Domino Effect: How cGMP/PKG Signaling Disarms β-Catenin
The beauty of this mechanism lies in its simplicity. In healthy colonocytes, APC maintains β-catenin at bay through proteasomal degradation. But when APC is mutated, β-catenin escapes regulation, translocates to the nucleus, and drives the transcription of genes such as cyclin D1, c-Myc, and survivin—a perfect recipe for malignancy.
By elevating intracellular cGMP, sulindac derivatives activate PKG, which phosphorylates β-catenin on specific residues, marking it for ubiquitination and proteasomal degradation. The downstream transcriptional program collapses, and the tumor cell loses its proliferative edge.
Beyond β-catenin, activated PKG also suppresses MAPK and RAS signaling, suggesting a broader anti-oncogenic network at play. The effect is both cytostatic and pro-apoptotic, making cGMP/PKG activation a versatile antitumor mechanism.
The Road to Translation: Opportunities and Caveats
From a drug development perspective, the advantages of these COX-free sulindac derivatives are clear:
- Safety: Elimination of COX inhibition sidesteps gastrointestinal and cardiovascular toxicity.
- Selectivity: Preferential targeting of PDE5/10 limits off-target effects.
- Efficacy: Dual pathway inhibition directly attacks tumor-promoting signaling networks.
However, translation to clinical reality faces familiar challenges. Optimizing oral bioavailability and pharmacokinetic stability remains critical, as many derivatives are rapidly metabolized. Furthermore, large-scale human trials are needed to verify whether the mechanisms observed in rodents and cell lines hold true in patients with FAP or sporadic adenomas.
Another hurdle is drug resistance. Tumor cells are notoriously adaptive, and long-term PDE inhibition might trigger compensatory signaling changes. Combination strategies—pairing sulindac derivatives with immunomodulators or targeted biologics—could mitigate this risk.
Looking Ahead: Redefining Chemoprevention in Oncology
If successful, this line of research could redefine what it means to “prevent” cancer. Instead of blunting inflammation, future chemopreventive drugs may restore disrupted signaling equilibrium, correcting molecular dysfunctions long before malignancy manifests.
By leveraging PDE5/10 inhibition, we could intervene not merely at the level of symptoms or pathways but at the genetic epicenter of carcinogenesis itself. This approach aligns with a broader movement in precision medicine—one that values targeted modulation over indiscriminate suppression.
In the story of sulindac, what began as an anti-inflammatory has evolved into a signal transduction modulator, bridging pharmacology and genetics in a way few drugs have before. It’s a testament to the power of scientific curiosity—and to the possibility that even old drugs can be reborn with a sharper purpose.
Conclusion
Colorectal cancer prevention is poised for a paradigm shift. The discovery that sulindac and its derivatives can suppress tumorigenesis through COX-independent mechanisms opens a new chapter in oncologic pharmacology. By targeting PDE5 and PDE10 to reactivate cGMP/PKG signaling, researchers have uncovered a path toward safer, more selective, and mechanistically intelligent therapy.
The implications reach far beyond colon cancer. cGMP signaling intersects with numerous cellular systems, suggesting that this strategy could apply to other malignancies marked by dysregulated β-catenin or MAPK activity.
For now, the journey from lab bench to clinic continues—but one thing is certain: the once-humble NSAID has matured into a sophisticated molecular tool, poised to outsmart cancer at its own game.
FAQ: Sulindac Derivatives and cGMP/PKG Signaling in Colorectal Cancer
1. Why develop sulindac derivatives without COX inhibition?
Because COX inhibition, while effective against inflammation, causes severe gastrointestinal and cardiovascular toxicity. Removing COX-binding allows researchers to retain sulindac’s anticancer properties—now attributed to PDE inhibition and cGMP/PKG activation—without endangering patients.
2. What makes PDE5 and PDE10 ideal cancer targets?
Both enzymes are overexpressed in colon tumors but absent in normal tissue. Their inhibition selectively elevates cGMP levels, reactivating PKG and promoting the degradation of oncogenic β-catenin—effectively reversing the downstream effects of APC mutations.
3. Could dual PDE5/10 inhibitors replace colon surgery in FAP?
Not yet, but that’s the vision. If proven safe and effective, such drugs could dramatically reduce adenoma burden, delay colectomy, and even serve as preventive agents for high-risk individuals with sporadic polyps.
