
Introduction
In the relentless war against cancer, the battlefield is not limited to tumor cells alone — it extends into the intricate signaling networks that sustain them. Among these molecular accomplices, cyclooxygenase-2 (COX-2) stands out as both a mediator and an amplifier of malignancy. Long recognized for its role in inflammation, COX-2 has emerged as a central orchestrator of tumor progression, angiogenesis, and drug resistance.
While conventional chemotherapy targets cell division directly, the strategic addition of COX-2 inhibitors introduces a subtler form of attack — one that dismantles the biochemical support systems tumors rely on for survival. This article explores, from a mechanistic standpoint, how COX-2 inhibition enhances the cytotoxic potential of chemotherapy, examining its effects on signaling pathways, apoptosis, angiogenesis, immune modulation, and multidrug resistance.
COX-2: The Inflammatory Conductor of Tumor Biology
The cyclooxygenase (COX) enzyme family includes two major isoforms: COX-1, a constitutive enzyme maintaining physiological homeostasis (e.g., gastric protection and platelet function), and COX-2, an inducible enzyme activated by inflammatory cytokines, oncogenes, and tumor promoters. In cancer, COX-2 expression is often upregulated 10- to 80-fold, particularly in malignancies such as colorectal, breast, prostate, pancreatic, and lung cancers.
The Prostaglandin Axis
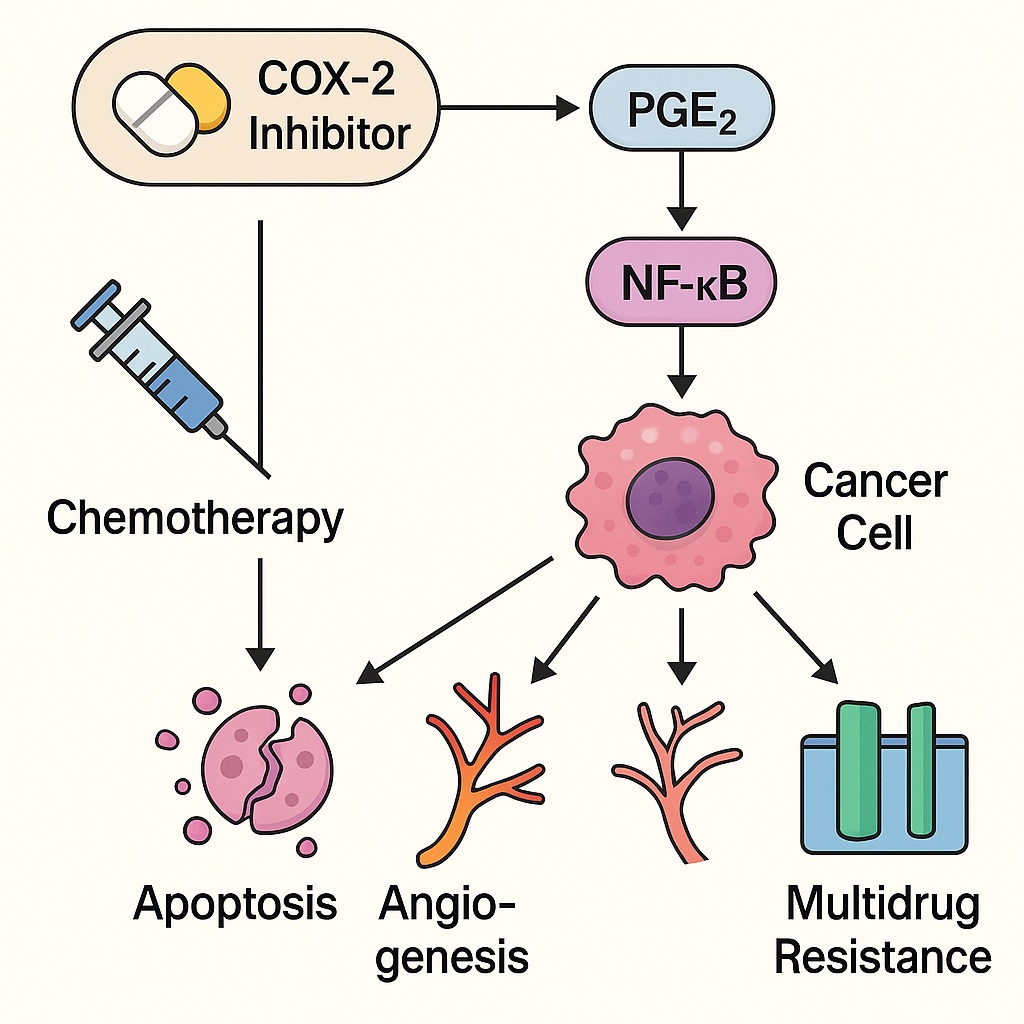
COX-2 catalyzes the conversion of arachidonic acid to prostaglandin H₂ (PGH₂), which is then transformed into various prostaglandins, notably prostaglandin E₂ (PGE₂). PGE₂ acts through four G-protein–coupled receptors (EP1–EP4), activating downstream pathways including PI3K/Akt, MAPK/ERK, and NF-κB, thereby promoting proliferation, survival, angiogenesis, and immune evasion.
This persistent inflammatory signaling does not merely accompany cancer — it fuels it. COX-2-derived prostaglandins increase vascular permeability, growth factor release, and cellular resistance to apoptosis, forming a biochemical shield around malignant tissue.
COX-2 and Chemoresistance
Increased COX-2 expression correlates with poor chemotherapy response. Mechanistically, PGE₂ signaling activates anti-apoptotic proteins such as Bcl-2 and survivin, suppresses caspase activation, and promotes the expression of multidrug resistance proteins (e.g., MDR1/P-glycoprotein). The result is a tumor microenvironment resilient to cytotoxic injury — one that sustains growth even under chemotherapeutic pressure.
The Molecular Rationale for Combining COX-2 Inhibitors with Chemotherapy
The fundamental concept of combination therapy is synergy — achieving therapeutic efficacy greater than the sum of its parts. COX-2 inhibitors, such as celecoxib, rofecoxib, and etoricoxib, potentiate chemotherapy by dismantling key biochemical defense systems within tumors. The major mechanistic advantages include:
- Sensitization of tumor cells to apoptosis
- Inhibition of angiogenesis
- Suppression of metastatic signaling
- Reduction of multidrug resistance
- Reprogramming of the tumor immune microenvironment
Let us explore each in molecular depth.
1. Reinstating Apoptosis: Disabling Cancer’s Survival Circuits
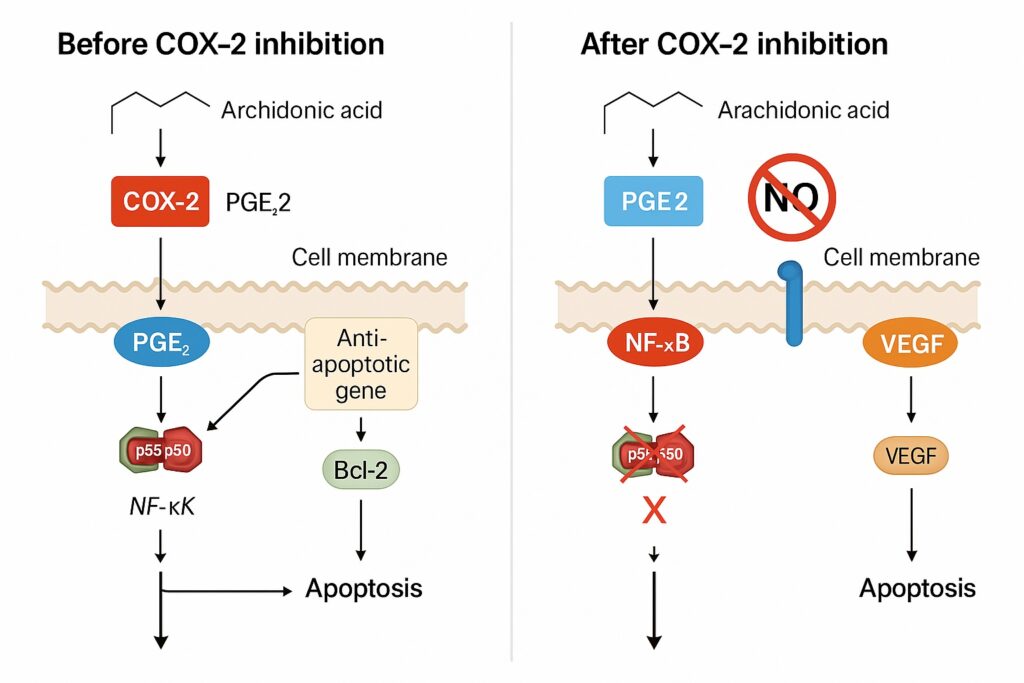
Apoptosis resistance is a defining hallmark of malignancy. Many chemotherapeutic agents — such as cisplatin, doxorubicin, and paclitaxel — rely on intrinsic (mitochondrial) or extrinsic (death receptor–mediated) apoptotic pathways to eliminate cancer cells. Unfortunately, COX-2–derived PGE₂ disrupts both.
COX-2, NF-κB, and Anti-Apoptotic Gene Expression
PGE₂ activates the NF-κB transcription factor, which upregulates Bcl-2, Bcl-xL, and XIAP — key inhibitors of caspase-dependent apoptosis. Chemotherapeutic agents that induce DNA damage are thus rendered less effective, as the apoptotic “execution” machinery is silenced.
COX-2 inhibitors counteract this effect by suppressing PGE₂ synthesis and thereby attenuating NF-κB signaling. This rebalances the intracellular environment toward pro-apoptotic dominance, restoring chemotherapy-induced cell death.
Studies show that celecoxib enhances cisplatin-induced apoptosis in non-small cell lung cancer by promoting caspase-9 activation and cytochrome c release — biochemical events normally blocked by COX-2 overexpression.
Downregulation of Survivin and Restoration of Caspase Cascade
Survivin, a member of the inhibitor of apoptosis (IAP) family, is a downstream target of PGE₂ signaling. COX-2 inhibition downregulates survivin expression, restoring mitochondrial permeability and enabling full caspase activation. The result is a sensitized apoptotic response, even in previously resistant tumor cells.
2. Suppressing Angiogenesis: Strangling the Tumor’s Lifeline
For tumors to grow beyond 1–2 mm in diameter, they must recruit new blood vessels. COX-2 is a potent promoter of angiogenesis, largely through PGE₂-mediated upregulation of vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF).
PGE₂ and Endothelial Activation
PGE₂ binds to EP2 and EP4 receptors on endothelial cells, activating cAMP/PKA and PI3K/Akt pathways. This results in endothelial proliferation, migration, and capillary tube formation. Chemotherapy alone cannot effectively suppress this process, as many agents trigger local hypoxia, which paradoxically induces VEGF expression.
COX-2 inhibition counterbalances this feedback, reducing both VEGF synthesis and microvessel density within tumors. In xenograft models of colon cancer, celecoxib combined with 5-fluorouracil (5-FU) decreased tumor microvessel density by nearly 60%, demonstrating angiostatic synergy.
Endothelial Sensitization to Chemotherapy
Endothelial cells are themselves partially resistant to chemotherapy. COX-2 inhibitors increase their susceptibility by reducing Bcl-2 expression and by impairing VEGF autocrine loops. This dual effect — anti-angiogenic and chemosensitizing — helps “starve” tumors both physically and biochemically.
3. Modulating the Tumor Microenvironment: From Pro-Inflammatory to Cytotoxic
Cancer is often described as a wound that never heals, perpetually trapped in a state of inflammation. Tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and regulatory T cells (Tregs) populate this environment, secreting cytokines that foster immune tolerance and angiogenesis.
COX-2 as a Master Regulator of Tumor Immunity
Through PGE₂ signaling, COX-2 skews macrophage polarization toward the M2 (immunosuppressive) phenotype, increases Treg infiltration, and suppresses cytotoxic CD8⁺ T-cell activity. It also induces IL-10 and TGF-β, further dampening anti-tumor immune responses.
By inhibiting COX-2, nonsteroidal anti-inflammatory drugs (NSAIDs) such as celecoxib and indomethacin can reprogram this microenvironment — reducing M2 macrophages, enhancing antigen presentation, and facilitating chemotherapy-induced immunogenic cell death.
In essence, COX-2 inhibition turns a tumor’s biochemical fortress into an inflammatory vulnerability.
4. Overcoming Multidrug Resistance (MDR)
Multidrug resistance is one of the most formidable barriers in oncology, often linked to overexpression of P-glycoprotein (P-gp) and multidrug resistance-associated proteins (MRPs) that actively efflux chemotherapeutic drugs out of tumor cells. COX-2 signaling contributes to MDR through PGE₂–EP4–PKA activation, which increases MDR1 gene transcription.
COX-2 Inhibitors as Modulators of Drug Efflux
Celecoxib and other selective COX-2 inhibitors have been shown to downregulate P-gp expression and inhibit its ATPase activity, leading to higher intracellular drug concentrations. In breast and colon carcinoma cell lines, the combination of celecoxib with doxorubicin resulted in a 3- to 5-fold increase in intracellular drug accumulation and enhanced cytotoxicity.
Synergistic Drug Accumulation
Moreover, COX-2 inhibition stabilizes tight junction integrity, reducing paracellular leakage and maintaining higher local drug concentrations in the tumor microenvironment. Thus, chemotherapy drugs are not only retained longer but also act more efficiently, translating into measurable gains in tumor regression.
5. Cell Cycle Control: Synchronizing Chemotherapy Timing
Many cytotoxic drugs act during specific cell cycle phases — for instance, paclitaxel during mitosis and 5-FU during the S-phase. COX-2-derived PGE₂ disrupts normal cell cycle checkpoints by upregulating cyclin D1 and CDK4, propelling tumor cells through the G₁/S transition prematurely.
COX-2 inhibition normalizes this cycle, increasing the proportion of cells in vulnerable phases during chemotherapy. This synchronization enhances the time-dependent efficacy of cytotoxic agents, a principle particularly useful in dose fractionation strategies.
6. COX-Independent Effects: Celecoxib’s Additional Molecular Targets
While most of celecoxib’s antitumor activity is attributed to COX-2 inhibition, several COX-independent pathways contribute to its synergy with chemotherapy.
Inhibition of the PI3K/Akt Pathway
Celecoxib directly inhibits phosphoinositide-dependent kinase-1 (PDK1), reducing downstream Akt phosphorylation. Since Akt signaling is pivotal in cell survival and chemoresistance, its inhibition amplifies chemotherapy-induced apoptosis. This effect is independent of prostaglandin synthesis and adds another dimension to celecoxib’s cytotoxic synergy.
Modulation of Endoplasmic Reticulum Stress
At higher concentrations, celecoxib triggers endoplasmic reticulum (ER) stress via the CHOP–GRP78 axis, leading to apoptotic cell death. In combination with drugs like paclitaxel or cisplatin, which also perturb protein folding, this results in additive ER stress–mediated lethality.
These COX-independent effects reinforce the rationale for celecoxib’s inclusion in multidrug regimens, particularly where resistance pathways converge on the PI3K/Akt network.
Pharmacological Considerations: Dosing, Selectivity, and Toxicity
The integration of COX-2 inhibitors into chemotherapy protocols demands careful pharmacologic balance. Selectivity, dose, and timing must be optimized to achieve synergy without amplifying toxicity.
Selective vs. Nonselective COX Inhibitors
Selective COX-2 inhibitors (coxibs) such as celecoxib, etoricoxib, and parecoxib provide superior tumor selectivity with fewer gastrointestinal side effects compared to traditional NSAIDs. However, their cardiovascular risk profile — related to prostacyclin suppression — necessitates vigilance, especially in long-term use.
Pharmacokinetic Compatibility
Celecoxib exhibits a half-life of 11 hours and achieves steady-state plasma levels conducive to continuous COX-2 suppression. When combined with chemotherapeutic agents like 5-FU, paclitaxel, or cisplatin, no significant pharmacokinetic interactions have been reported, though both hepatic metabolism and renal clearance warrant monitoring.
Optimizing Treatment Schedule
Experimental data suggest sequential administration — COX-2 inhibitor pre-treatment followed by chemotherapy — yields superior outcomes compared to simultaneous dosing. Pre-inhibition of COX-2 downregulates anti-apoptotic defenses, priming tumor cells for subsequent cytotoxic assault.
Integrating Molecular Insight into Clinical Translation
While molecular data convincingly support COX-2 inhibition as a chemosensitizing strategy, clinical outcomes have been more nuanced. Variability in tumor COX-2 expression, genetic polymorphisms in prostaglandin synthase genes, and differences in drug metabolism affect response.
Predictive Biomarkers
COX-2 expression levels, PGE₂ serum concentrations, and EP receptor profiling are emerging as predictive biomarkers for selecting patients likely to benefit from combination therapy. Additionally, polymorphisms in CYP2C9, which metabolizes celecoxib, may influence efficacy and toxicity, warranting personalized dosing strategies.
Toward Precision Chemotherapy
Future protocols may integrate COX-2 inhibition into precision oncology frameworks, pairing molecular diagnostics with targeted anti-inflammatory modulation. The ultimate goal is to transform COX-2 inhibitors from adjuvant drugs into mechanism-based amplifiers of chemotherapeutic precision.
The Broader Implications: Rethinking Cancer as an Inflammatory Disease
The success of COX-2 inhibitors in modulating tumor response underscores a deeper conceptual shift: cancer is not merely a genetic disease but an inflammatory ecosystem. The persistent activation of COX-2 and downstream PGE₂ signaling blurs the line between wound healing and tumor progression.
Chemotherapy, traditionally viewed as a weapon of brute cytotoxicity, gains sophistication when paired with molecular disarmament of inflammatory pathways. The combination embodies a duality of attack — direct cytolysis of tumor cells and concurrent suppression of the microenvironment that nurtures them.
Thus, COX-2 inhibitors are not auxiliary agents but strategic allies — transforming chemotherapy from a blunt force into a more intelligent, multi-axis offensive.
Conclusion
The combination of chemotherapy with COX-2 inhibition represents a paradigm shift in oncologic pharmacology. By dismantling the biochemical scaffolding of tumor survival — including apoptosis resistance, angiogenesis, and immune evasion — COX-2 inhibitors render malignancies more vulnerable to standard cytotoxic therapy.
Mechanistically, this synergy unfolds through:
- Suppression of PGE₂–NF-κB–Bcl-2 signaling, reinstating apoptosis
- Inhibition of VEGF-driven angiogenesis, limiting nutrient supply
- Reduction of multidrug resistance gene expression, enhancing drug retention
- Reprogramming of the tumor microenvironment, improving immune surveillance
- COX-independent modulation of Akt and ER stress pathways, broadening cytotoxic reach
While clinical translation remains tempered by cardiovascular concerns and tumor heterogeneity, the molecular foundation is compelling. The future of this strategy lies in precision targeting, biomarker-guided therapy, and rational sequence design.
In the complex symphony of cancer treatment, COX-2 inhibitors do not replace chemotherapy — they tune it, refine it, and restore its potency by silencing the tumor’s inflammatory orchestra.
FAQ: COX-2 Inhibitors and Chemotherapy
1. How do COX-2 inhibitors make chemotherapy more effective?
They suppress prostaglandin E₂ signaling, which drives anti-apoptotic, angiogenic, and multidrug resistance pathways. This re-sensitizes tumor cells to chemotherapy and enhances cytotoxic efficiency.
2. Are the effects of COX-2 inhibitors purely anti-inflammatory?
No. In addition to blocking prostaglandin synthesis, agents like celecoxib also inhibit PI3K/Akt signaling, induce endoplasmic reticulum stress, and directly affect tumor metabolism — providing COX-independent anticancer benefits.
3. Can COX-2 inhibitors be used for all cancer patients?
Not universally. Patients with high cardiovascular risk or low tumor COX-2 expression may derive less benefit. Molecular profiling and careful clinical monitoring are essential before inclusion in combination protocols.