

Introduction: A Meeting Between Metabolism and Mechanism
At first glance, metformin and sildenafil belong to different universes of pharmacotherapy. One is the cornerstone of modern diabetes management, celebrated for its humble ability to curb hepatic glucose output and sensitize cells to insulin. The other—sildenafil citrate—entered fame through entirely different pathways of physiology, where it revolutionized the treatment of erectile dysfunction by liberating nitric oxide–mediated vasodilation.
Yet, in the intertwined reality of modern medicine, these two drugs often coexist. Diabetic men, a population commonly prescribed metformin, are also disproportionately affected by erectile dysfunction—a condition for which sildenafil remains the most prescribed remedy. This therapeutic overlap prompts a fundamental question: Do these two drugs interfere with each other once inside the body?
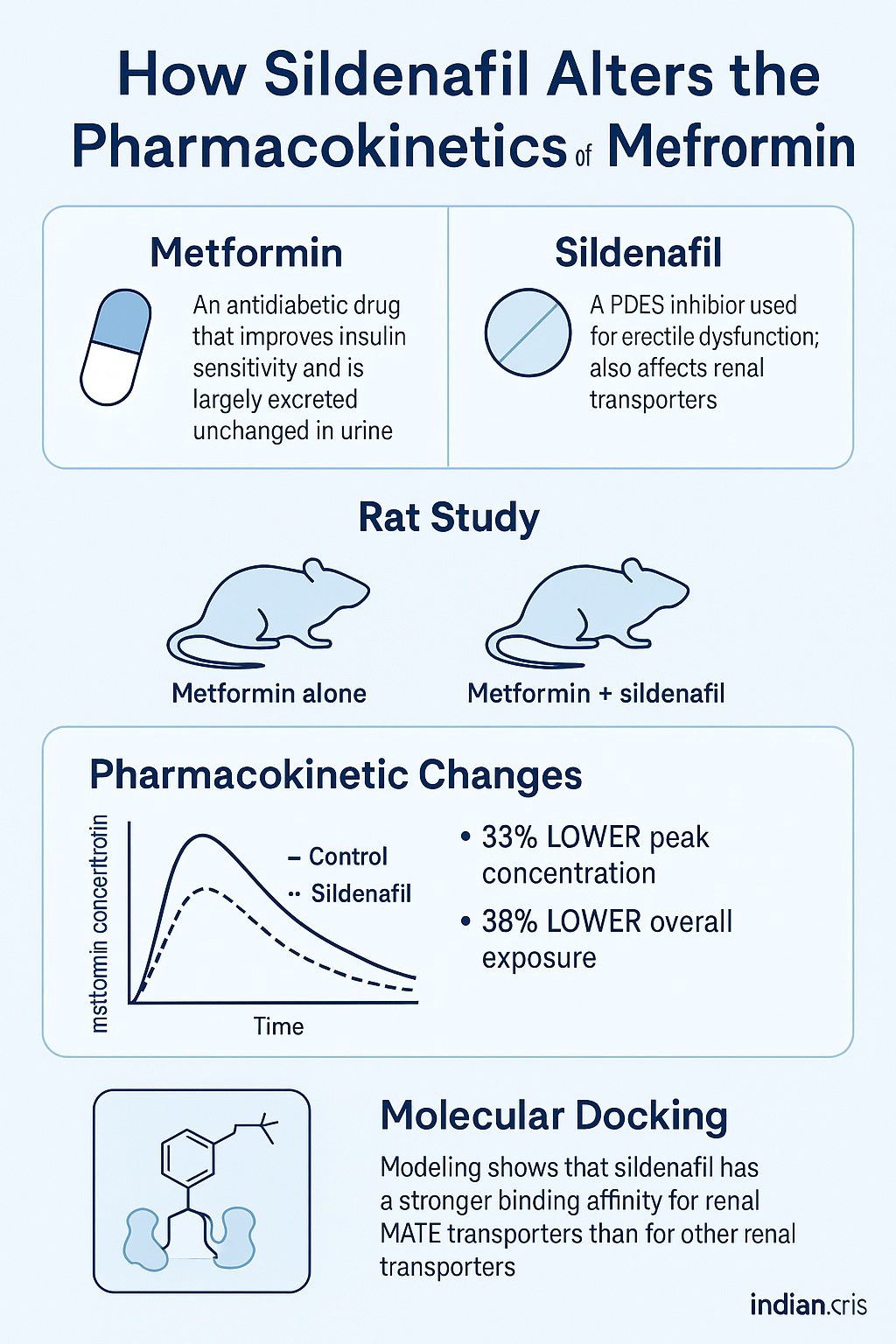
In a recent and elegant study published in the Indian Journal of Pharmacology (2024), Dewani and colleagues explored precisely this question. Using a dual approach that combined in vivo pharmacokinetic studies in rats and in silico molecular docking analyses, they investigated whether sildenafil modifies how metformin is absorbed, distributed, or cleared. Their conclusion was striking: sildenafil does indeed alter the pharmacokinetics of metformin, most likely through its interaction with renal transporters responsible for the drug’s elimination.
Metformin: The Tireless Workhorse of Glucose Regulation
Metformin hydrochloride, a biguanide derivative, remains the first-line pharmacologic agent in type 2 diabetes mellitus (T2DM). Its mechanisms are multifaceted—decreasing hepatic gluconeogenesis, enhancing peripheral glucose uptake, and slightly inhibiting intestinal absorption of glucose. Unlike most drugs, metformin is not metabolized; it is excreted unchanged in urine via active tubular secretion mediated primarily by two transporter families: organic cation transporters (OCTs) and multidrug and toxin extrusion proteins (MATEs).
This dependence on renal transporters makes metformin pharmacokinetics particularly vulnerable to drug–drug interactions. Inhibitors of OCT or MATE—such as cimetidine or proton-pump inhibitors—are known to reduce metformin clearance, leading to elevated plasma concentrations and, in extreme cases, lactic acidosis.
However, the current investigation by Dewani et al. revealed something counterintuitive: rather than increasing metformin exposure, sildenafil reduced its maximum plasma concentration (Cmax) and overall exposure (AUC). This suggested a different kind of interaction—one that might involve transporter competition at the level of renal elimination rather than absorption.
Sildenafil: Beyond the Blue Pill
Sildenafil citrate, a selective phosphodiesterase type 5 (PDE5) inhibitor, is rapidly absorbed after oral administration, reaching peak plasma concentrations within an hour and demonstrating about 40% bioavailability. It is metabolized in the liver via the CYP3A4 and CYP2C9 pathways to its active metabolite, N-desmethyl sildenafil.
While the world knows sildenafil for its vasodilatory prowess in erectile tissues, few realize that PDE5 enzymes are distributed widely across vascular, cardiac, and renal tissues. This systemic presence allows sildenafil to exert subtle modulatory effects on organ perfusion, including the kidney—an organ that happens to be metformin’s primary route of elimination.
Thus, the study’s central hypothesis emerged naturally: could sildenafil, by binding to renal transporters or altering renal hemodynamics, influence metformin’s pharmacokinetics?
Methodological Overview: Rats, HPLC, and Docking
The study was a model of pharmacological precision. Sixteen male Sprague–Dawley rats were divided into two groups:
- Control group: received metformin alone (200 mg/kg orally)
- Experimental group: received metformin (200 mg/kg) plus sildenafil (2.5 mg/kg)
Blood samples were collected at multiple intervals up to 7 hours post-dose, and plasma concentrations were quantified using a validated high-performance liquid chromatography (HPLC) method with UV detection at 224 nm. The extraction relied on solid-phase extraction (SPE) with Strata-X cartridges, achieving over 85% recovery for both drugs.
Importantly, the team also performed molecular docking using the crystal structures of OCT1 and MATE (Protein Data Bank ID: 5XJJ) to simulate sildenafil’s potential interactions with these transporters. This dual strategy—analytical and computational—allowed the authors to link pharmacokinetic alterations to probable molecular mechanisms.
The Pharmacokinetic Outcome: Sildenafil Changes the Metformin Curve
The results were unambiguous. When co-administered with sildenafil, metformin exhibited a 33% lower Cmax and a 38% smaller AUC, while its half-life (t½) decreased by about 0.3 hours compared to control rats.
To a pharmacologist, these numbers carry weight. They indicate that sildenafil either reduced metformin absorption or enhanced its elimination. Since metformin’s absorption is slow, saturable, and largely independent of metabolism, the latter explanation is more plausible.
Interestingly, the apparent volume of distribution (Vd) increased from 0.21 L in controls to 0.31 L in sildenafil-treated animals, while the elimination constant (Ke) also rose (0.417 vs. 0.35 h⁻¹). Both shifts are consistent with accelerated clearance, potentially through upregulated or competitively modified transporter activity.
In practical terms, the combination led to less metformin staying in circulation for a shorter time—a pharmacokinetic footprint that may have clinical implications for glycemic control in diabetic patients who use sildenafil chronically.
The Molecular Mystery: How Docking Revealed a Transporter Story
To uncover the underlying mechanism, the team conducted in silico docking of sildenafil against the two key metformin transporters: OCT1 (responsible for hepatic uptake) and MATE1/2-K (involved in renal excretion).
The results were revealing. Sildenafil exhibited a stronger binding affinity for the MATE transporter (binding energies around −9.5 kcal/mol) compared to OCT1 (−7.3 kcal/mol). Hydrogen bonding and hydrophobic interactions with MATE residues—particularly THR70, ASN74, and TRP266—suggested stable and energetically favorable complex formation.
In contrast, its interaction with OCT1 was weak, involving a single hydrogen bond and no significant hydrophobic contacts.
These findings imply that sildenafil might occupy or alter MATE binding sites, thereby modifying how metformin is extruded from renal tubular cells into urine. The observed decrease in plasma exposure aligns with a scenario in which metformin is cleared more efficiently, or alternatively, redistributed due to altered transporter kinetics.
Why Transporters Matter: The Pharmacokinetic Gatekeepers
Drug transporters like OCT1, OCT2, and MATE1 are increasingly recognized as key determinants of pharmacokinetic behavior. They govern how drugs move across cellular membranes—whether entering hepatocytes for metabolism or leaving renal cells for excretion.
Metformin’s dependence on these transporters is near absolute; it lacks lipophilicity to cross membranes by passive diffusion. Hence, any molecule that interferes with these proteins—either by inhibition or competitive binding—can profoundly affect metformin’s pharmacokinetics.
Sildenafil’s newfound interaction with MATE proteins positions it as a potential pharmacokinetic modulator rather than a mere PDE5 inhibitor. While this may sound esoteric, in clinical settings it could translate to altered glycemic profiles, reduced efficacy, or unpredictable drug levels in diabetic patients.
Analytical Rigor: Validating the HPLC Approach
Before interpreting pharmacokinetic shifts, the researchers ensured their analytical platform was robust. The developed HPLC method was validated according to ICH guidelines, meeting criteria for selectivity, linearity, precision, and accuracy.
The method showed excellent linearity (r² = 0.976) over 5–2000 ng/mL and maintained inter- and intra-day precision with relative standard deviation (%RSD) below 6%. Recovery rates hovered between 85–91% for metformin and 87–90% for sildenafil.
Notably, the method’s limit of quantification reached 8 ng/mL for metformin and 10 ng/mL for sildenafil—demonstrating sensitivity suitable for low plasma concentrations. The retention time under isocratic conditions was under five minutes, making the technique rapid and cost-effective for pharmacokinetic applications.
This attention to analytical detail strengthens confidence that the pharmacokinetic variations observed were truly biological and not artifacts of measurement.
A Broader Context: Polytherapy, Diabetes, and the Risk of Hidden Interactions
Polypharmacy is an inevitable companion of chronic disease management. Diabetic patients frequently receive not only antihyperglycemics but also antihypertensives, lipid-lowering agents, and, for many men, PDE5 inhibitors.
Drug–drug interactions, particularly those mediated by transporters, remain a largely underappreciated hazard. While most clinicians are attuned to cytochrome P450 interactions, transporter-mediated effects—like the MATE–sildenafil interplay—often go unnoticed until clinical outcomes deviate from expectation.
For example, diminished metformin exposure could subtly compromise glycemic control, leading to therapeutic “mystery failures.” Conversely, in some individuals with impaired renal function, transporter inhibition could paradoxically increase metformin retention, risking lactic acidosis.
Thus, understanding these mechanisms is not just an academic exercise—it is a matter of optimizing patient safety and treatment efficacy.
Pharmacological Implications: From Bench to Bedside
While this study was conducted in rats, its implications stretch toward clinical pharmacology. If similar effects occur in humans, the co-administration of sildenafil and metformin might require dose adjustments or at least therapeutic monitoring.
A reduced metformin AUC could mean lower intracellular concentrations in target tissues, potentially diminishing its insulin-sensitizing action. For patients already struggling with glycemic control, even a modest pharmacokinetic shift could tilt the balance.
Furthermore, these findings highlight a broader principle: non-metabolic drugs can still profoundly affect each other via transporter-mediated crosstalk. As precision medicine advances, incorporating transporter profiling into drug interaction studies will become increasingly essential.
The Role of Molecular Docking in Pharmacology
One of the strengths of this investigation lies in its integration of computational chemistry. Molecular docking provided visual and quantitative insight into how sildenafil might fit within the active sites of transport proteins.
Using PyRx for screening and BIOVIA Discovery Studio for visualization, the researchers simulated the spatial orientation, binding residues, and interaction energies. The high-affinity docking with MATE (PDB ID 5XJJ) reinforced the in vivo pharmacokinetic findings, bridging experimental observation with molecular plausibility.
This dual analytical–molecular approach represents a gold standard in modern pharmacokinetic research, where in silico modeling guides and contextualizes in vivo results.
Limitations and Future Directions
No scientific story is complete without caveats. The study’s animal model provides a controlled system but may not fully replicate human transporter expression or pharmacodynamics. Metformin’s pharmacokinetics in humans are influenced by genetic polymorphisms in OCT and MATE genes—factors that could modify how sildenafil interacts in clinical settings.
Moreover, the study focused on acute, single-dose administration. Chronic co-administration may produce adaptive changes in transporter expression or renal perfusion, altering the interaction profile.
Future work should:
- Validate these findings in human subjects or transgenic mice expressing human transporters.
- Explore whether sildenafil metabolites share similar binding affinities.
- Investigate potential interactions with other PDE5 inhibitors like tadalafil or vardenafil.
Such research could define whether this phenomenon is a sildenafil-specific effect or a class-wide property of PDE5 inhibitors.
Conclusion: A Subtle Interaction With Significant Meaning
The investigation by Dewani and colleagues elegantly demonstrates that even well-characterized drugs can surprise us. Sildenafil, the emblem of vascular pharmacology, crosses paths with metformin, the workhorse of metabolic control, in a dance choreographed by renal transporters.
Through meticulous HPLC quantification and sophisticated molecular docking, the study established that sildenafil reduces metformin plasma exposure by influencing MATE-mediated elimination. This interaction—though moderate—adds a valuable piece to the puzzle of transporter-mediated drug interactions in metabolic disease management.
In clinical translation, the message is clear: monitoring, awareness, and mechanistic understanding remain vital when combining medications that share renal or transporter pathways. What began as a study on rats may ultimately refine how physicians prescribe for millions of diabetic men worldwide.
FAQ: Sildenafil–Metformin Interaction
1. Why does sildenafil affect metformin’s pharmacokinetics?
Sildenafil interacts with renal MATE transporters, which are responsible for pumping metformin from kidney cells into urine. This interaction alters metformin’s clearance, reducing its plasma levels and exposure time.
2. Does this interaction pose a health risk to diabetic patients?
In most cases, it’s unlikely to cause acute toxicity, but it could modestly reduce metformin’s therapeutic efficacy. Patients on chronic combination therapy should be monitored for changes in blood glucose control.
3. Is this effect unique to sildenafil, or do other PDE5 inhibitors do the same?
The study focused solely on sildenafil, but since other PDE5 inhibitors share structural similarities, they might exhibit comparable interactions. Further studies are needed to confirm whether this is a class effect.
