Introduction
In modern pharmacology, a small set of enzymes have managed to transform the way physicians view vascular physiology, sexual health, and cardiopulmonary medicine. Among these, phosphodiesterase 5 (PDE5) holds a particularly prominent role. Once considered a niche enzyme of limited clinical interest, PDE5 rose to global fame with the advent of sildenafil—better known to the public as Viagra. What began as a serendipitous discovery for erectile dysfunction (ED) has evolved into a vast field of therapeutic exploration, ranging from pulmonary arterial hypertension to oncology.
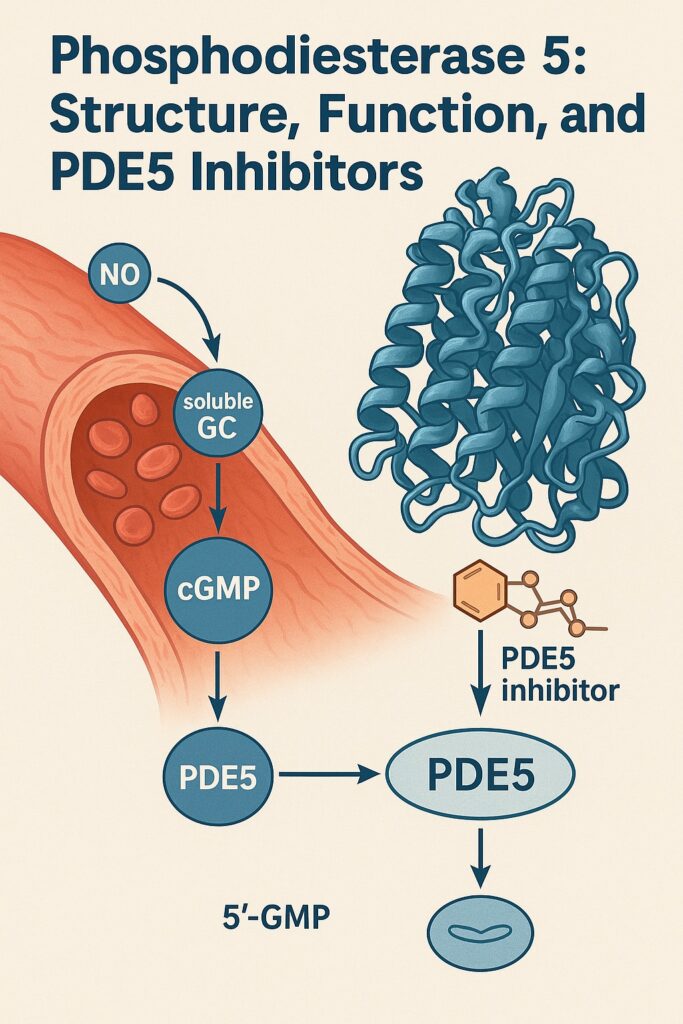
The importance of PDE5 stems from its role in regulating cyclic guanosine monophosphate (cGMP), the critical second messenger in nitric oxide (NO)–mediated vasodilation. By controlling the degradation of cGMP, PDE5 essentially acts as the “brake pedal” for smooth muscle relaxation. Inhibiting PDE5 therefore amplifies nitric oxide signaling, prolongs vasodilation, and enhances tissue perfusion. Such a seemingly simple mechanism belies the enormous physiological and clinical consequences that follow.
This article explores the structural biology of PDE5, its regulation, the pharmacological mechanisms of its inhibitors, and the growing repertoire of therapeutic applications. While the public associates PDE5 inhibitors almost exclusively with sexual medicine, the science reveals a much broader narrative—one that continues to unfold with each passing decade.
Structural Features of PDE5
Understanding PDE5 requires a dive into its molecular anatomy. PDE5 belongs to the broader family of phosphodiesterases, enzymes responsible for hydrolyzing cyclic nucleotides (cAMP and cGMP). PDE5 is selective for cGMP and is widely expressed in vascular smooth muscle, platelets, and the corpus cavernosum of the penis.
Structurally, PDE5 consists of two major domains: an N-terminal regulatory domain and a C-terminal catalytic domain. The regulatory region contains GAF (cGMP-binding PDE, Anabaena adenylyl cyclase, and E. coli FhlA) domains, which bind cGMP and control enzyme conformation. These GAF domains act like allosteric “sensors,” fine-tuning the enzyme’s activity based on intracellular signaling.
The catalytic domain, in contrast, houses the active site where hydrolysis of cGMP occurs. Crystallographic studies have revealed a deep hydrophobic pocket lined with residues that interact with the purine ring of cGMP. A zinc ion and a magnesium ion are essential cofactors for catalysis, stabilizing the phosphate backbone during hydrolysis. Importantly, PDE5 inhibitors mimic cGMP’s structure and lodge themselves within this catalytic pocket, effectively blocking enzymatic activity.
Such structural precision explains why even subtle differences in inhibitor design—whether sildenafil, tadalafil, or vardenafil—translate into distinct pharmacokinetic profiles and tissue selectivity. In many ways, PDE5’s architecture serves as both its strength and its vulnerability: a tightly regulated enzyme whose Achilles’ heel can be exploited by small molecules with clinical benefit.
Regulation of PDE5 Activity
Biological systems thrive on feedback loops, and PDE5 is no exception. Its activity is modulated by both allosteric binding and phosphorylation.
When intracellular cGMP levels rise, cGMP binds to the GAF domains of PDE5, inducing conformational changes that increase catalytic efficiency. This forms a negative feedback loop: more cGMP leads to greater PDE5 activity, which in turn accelerates cGMP breakdown. Such self-regulation prevents runaway vasodilation and maintains vascular tone within safe limits.
Another layer of control comes from phosphorylation by protein kinase G (PKG). When activated by cGMP, PKG phosphorylates PDE5 at serine residues, further enhancing its catalytic activity. This ensures that as nitric oxide signaling escalates, PDE5 rapidly engages to keep vascular reactivity under control.
This tight regulation is physiologically advantageous but also explains why pharmacological inhibition is so potent. By interrupting these natural braking mechanisms, PDE5 inhibitors shift the vascular balance dramatically toward relaxation. Clinically, this translates into improved penile erections, reduced pulmonary arterial pressures, and potentially improved tissue oxygenation in diverse organs.
Pharmacology of PDE5 Inhibitors
The modern era of PDE5 inhibitors began in the early 1990s, when sildenafil was developed as a potential anti-anginal agent. Its failure in ischemic heart disease trials turned into triumph when participants reported improved erections—a side effect too significant to ignore. By 1998, sildenafil was approved as the first oral therapy for erectile dysfunction, ushering in a revolution in sexual medicine.
Pharmacologically, PDE5 inhibitors share a common mechanism: they compete with cGMP for binding at the catalytic site of PDE5, preventing its hydrolysis. The result is prolonged cGMP activity, greater smooth muscle relaxation, and enhanced vasodilation. Yet each agent differs in chemical structure, potency, and half-life.
- Sildenafil: half-life 3–5 hours; effective for erectile dysfunction and pulmonary hypertension.
- Tadalafil: half-life ~17 hours; provides longer therapeutic windows, sometimes termed the “weekend pill.”
- Vardenafil: similar to sildenafil but with faster onset and slightly greater potency.
- Avanafil: newer generation, highly selective for PDE5 with rapid onset.
Selectivity is critical. Off-target inhibition of other PDE isoforms can lead to unwanted effects: inhibition of PDE6 in the retina explains sildenafil-induced visual disturbances, while PDE11 inhibition by tadalafil may cause back pain and myalgia. Despite these nuances, the overall safety profile of PDE5 inhibitors is remarkably favorable given their widespread use.
Clinical Applications Beyond Erectile Dysfunction
Although PDE5 inhibitors became household names due to sexual health, their clinical portfolio has expanded dramatically. Today, they are recognized as versatile agents across multiple disciplines.
Pulmonary Arterial Hypertension (PAH)
PAH is characterized by elevated pulmonary vascular resistance, progressive right heart failure, and high mortality. Sildenafil and tadalafil are both approved for PAH, where they reduce pulmonary pressures, improve exercise tolerance, and enhance quality of life. By enhancing nitric oxide signaling in pulmonary vasculature, PDE5 inhibitors directly counteract the vasoconstrictive pathology of PAH.
Benign Prostatic Hyperplasia (BPH)
Lower urinary tract symptoms in BPH have traditionally been treated with alpha-blockers and 5-alpha-reductase inhibitors. However, tadalafil is now approved for symptomatic BPH, reflecting its ability to relax smooth muscle in the prostate and bladder neck. This dual utility—improving both urinary symptoms and erectile function—has made tadalafil particularly attractive in middle-aged men.
Cardiac Conditions
Emerging research suggests that PDE5 inhibitors may benefit conditions like systolic heart failure, left ventricular hypertrophy, and ischemia-reperfusion injury. By improving myocardial oxygen delivery and attenuating maladaptive remodeling, PDE5 inhibition offers cardioprotective potential. Clinical translation, however, remains in early stages.
Oncology and Other Fields
Intriguingly, PDE5 inhibitors have been investigated as adjuncts in oncology, where they may enhance tumor perfusion, sensitize tumors to chemotherapy, and modulate immune responses. Animal studies also point toward benefits in neurodegenerative disorders, given PDE5’s role in neuronal signaling. While these remain experimental, they underscore the enzyme’s far-reaching physiological impact.
Safety Profile and Adverse Effects
No discussion of PDE5 inhibitors is complete without acknowledging their side effects. Fortunately, these are generally mild and predictable.
The most common include headache, flushing, nasal congestion, dyspepsia, and dizziness—direct consequences of systemic vasodilation. Visual disturbances such as blue-tinged vision are linked to cross-inhibition of retinal PDE6. Tadalafil’s musculoskeletal complaints likely stem from PDE11 inhibition.
Serious adverse events are rare but important. Concomitant use with nitrates can precipitate life-threatening hypotension, a contraindication enshrined in every prescribing guideline. Caution is also advised in men with unstable cardiovascular disease. Rare reports of priapism, sudden hearing loss, or optic neuropathy exist, though causal links are debated.
Overall, when prescribed appropriately, PDE5 inhibitors have a favorable risk–benefit ratio. Their global usage attests to their safety, with millions of doses consumed each year without widespread harm.
The Future of PDE5 Research
Despite decades of clinical use, PDE5 research is far from exhausted. Several promising directions warrant attention.
First, novel inhibitors with greater selectivity and improved pharmacokinetics are under development. The aim is to retain therapeutic efficacy while minimizing side effects. Avanafil is one example of this newer generation, offering rapid onset with reduced retinal effects.
Second, repurposing PDE5 inhibitors remains an active field. Their vasodilatory, anti-inflammatory, and immunomodulatory properties may prove beneficial in diseases as diverse as Alzheimer’s disease, systemic sclerosis, and even certain cancers.
Third, personalized medicine may refine PDE5 inhibitor therapy. Genetic polymorphisms in PDE5 or nitric oxide signaling pathways could explain inter-individual variability in response. Identifying such markers could guide more tailored treatment strategies.
Finally, the COVID-19 pandemic reignited interest in PDE5 inhibitors due to their potential in ameliorating pulmonary vascular dysfunction. While data remain preliminary, such explorations highlight how PDE5 inhibitors continue to surprise, decades after their introduction.
Conclusion
Phosphodiesterase 5 is more than just the molecular target of a famous blue pill. It is a finely tuned enzyme central to vascular physiology, with inhibitors that have reshaped therapeutic strategies across cardiology, pulmonology, urology, and beyond.
The journey of PDE5 inhibitors underscores the serendipity of drug discovery—what began as a failed angina therapy became a revolution in sexual health, and later, a lifeline for patients with pulmonary hypertension. As new indications emerge, PDE5 inhibitors remind us that molecular biology is rarely linear, and clinical innovation often comes from unexpected pathways.
Ultimately, the story of PDE5 reflects the best of translational medicine: from bench crystallography to bedside rehabilitation, from molecules to meaning, from structure to salvation.
FAQ
1. Why do PDE5 inhibitors improve erections?
They block the enzyme that breaks down cGMP in penile tissue. By prolonging cGMP signaling, smooth muscle relaxes, arterial inflow increases, and the penis achieves and maintains erection more effectively.
2. Are PDE5 inhibitors safe for long-term use?
Yes, in most patients. Millions worldwide use them chronically for erectile dysfunction or pulmonary hypertension. Side effects are generally mild, but contraindications—especially concurrent nitrate therapy—must be respected.
3. Can PDE5 inhibitors treat diseases beyond ED and PAH?
Potentially. Research is exploring applications in heart failure, benign prostatic hyperplasia, neurodegeneration, and oncology. While not yet routine, the expanding evidence suggests PDE5 inhibitors may play roles well beyond their original scope.